In a recent study posted to the medRxiv* pre-print server, scientists at nference Labs investigated the origin of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) new variant of concern (VOC) Omicron's unique spike (S) gene insertion ins214EPE. It is an in-frame insertion of nine nucleotides between positions 22204 and 22207, constituting a ribonucleic acid (RNA) loop which is more prone to insertions than an RNA stem.
 Study: On the origin of Omicron's unique Spike gene insertion. Image Credit: Naeblys / Shutterstock
Study: On the origin of Omicron's unique Spike gene insertion. Image Credit: Naeblys / Shutterstock
Background
Compared to other SARS-CoV-2 VOCs, Omicron S has 26 distinct amino acid mutations, including two deletions, one insertion, and 23 substitutions. Only the insertion mutation, ins214EPE, has not been previously identified in any SARS-CoV-2 lineage. Hence, characterizing Omicron's mutational profile is essential for interpreting its distinct clinical phenotype. It is possible that ins214EPE impacts the transmissibility or infectivity of Omicron; moreover, it might point out whether SARS-CoV-2 could be exploiting human cells as an 'evolutionary sandbox' to produce new variants.
About the study
In the present study, researchers obtained core mutations of each SARS-CoV-2 parental lineages, including Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Omicron (BA.1), and Delta (B.1.617.2) VOCs, Lambda (C.37), Mu (B.1.621), Eta (B.1.525), Iota (B.1.526), and Kappa (B.1.617.1) variants of interest (VOIs) from the coronavirus antiviral research database (CoV-RDB) database.
Likewise, the team retrieved all SARS-CoV-2 genomes corresponding to Omicron directly from the global initiative on sharing all influenza data (GISAID) databases. The team also used the genomes from the GISAID database and epidemiology data from Our World in Data (OWID) to identify surge-associated mutations. These mutations are present in at least 100 SARS-CoV-2 sequences in the GISAID database; moreover, their prevalence increases at the same rate as reverse transcription-polymerase chain reaction (RT-PCR) positivity in any given country.
Further, the researchers performed a 9-mer nucleotide search to identify candidate viral and human templates for ins214EPE. They searched the Gencode database for human transcriptomes, the GISAID database for SARS-CoV-2 genomes, and the National Center for Biotechnology Information (NCBI) database for Coronaviridae viruses.
Furthermore, the team assessed homology between 35 nucleotide regions flanking EPE insertion and origin sites of all candidate templates using biopython v1.76. The Bio.pairwise2 module of the biopython software performs a global alignment of nucleotide sequence using a custom scoring scheme, with a score range from zero (no match) to 175 (perfect match). Lastly, they defined a Normalized Homology Score (NHS), ranging from 0 to 100, to assess the homology between shorter upstream and downstream sequences equivalent to seven nucleotides.
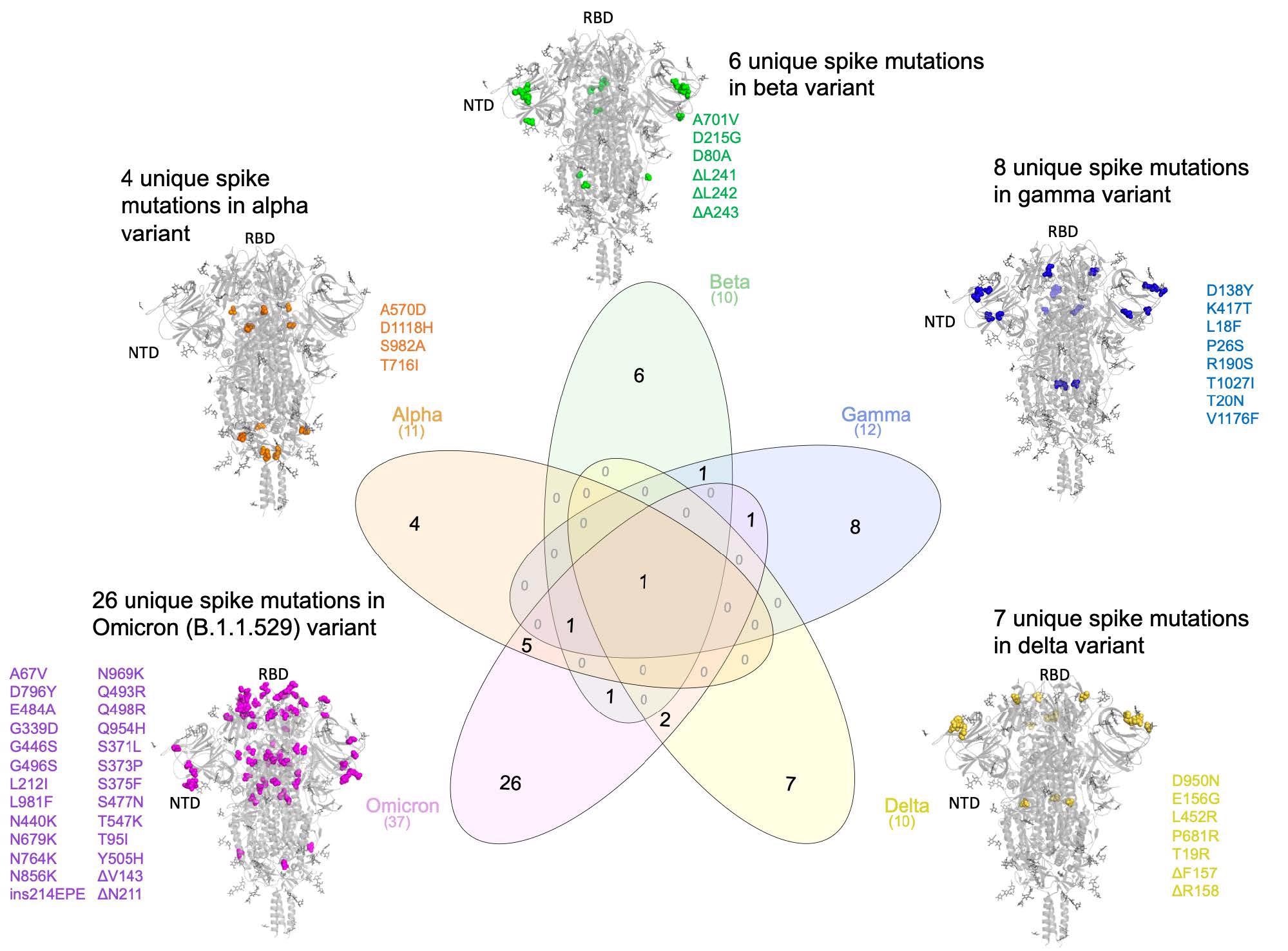 Comparing the lineage-specific Spike protein mutations in the SARS-CoV-2 variants of concern
Comparing the lineage-specific Spike protein mutations in the SARS-CoV-2 variants of concern
Study findings
The authors identified 16 surge-associated mutations in the Omicron VOC. Omicron had 26 unique mutations compared to other VOCs, while it shared seven of these mutations with the Alpha VOC. The authors compared these mutations to mutations from 5,781,715 genomes corresponding to ~1500 lineages from the GISAID database. The search yielded a specific, novel insertion mutation ins214EPE in Omicron, previously not observed in any SARS-CoV-2 lineage. A total of 1168 SARS-CoV-2 genomes in GISAID harbored ins214EPE, of which 1164 were classified as Omicron.
Further, the authors found template switching as a plausible mechanism for the origin of ins214EPE in Omicron. In the Coronaviridae family, template switching is a normal part of the life cycle responsible for subgenomic RNAs (sgRNAs) synthesis. However, the ins214EPE mutation in Omicron was unique in having exactly nine nucleotides, which made it a borderline case between short and long insertional mutations. Moreover, it did not use uracil nucleotides and was monophyletic. Most importantly, the EPE insertion occurred near previously known sites of potential non-canonical template switching.
Surprisingly, all the candidate templates generally had the NHS distributions similar to the distribution observed previously for randomly selected SARS-CoV-2 35-mers. Accordingly, the candidates with the highest degrees of homology in the flanking upstream or downstream sequences were from the SARS-CoV-2 lineage B.1.609 and AY.103, the HCoV-229E S protein, and human transcripts of actinin, alpha 1 (ACTN1) and endoplasmic reticulum membrane protein complex 4 (EMC4), with NHS scores of 69, 66, 63, 74, and 71, respectively. The reverse complement of the human transmembrane protein 245 (TMEM245) transcript showed more homology in shorter sequences directly upstream and downstream of the EPE insertion.
The Ins214EPE insertion mapped to the N-terminal domain (NTD) of Omicron S, close to the site of a known human T-cell epitope on SARS-CoV-2. Hence, further research should investigate whether ins214EPE insertion mutation could help SARS-CoV-2 escape T-cell immunity. Additionally, it is vital to understand its functional significance and evolutionary origin in Omicron, as the PRRA insertion in the original SARS-CoV-2 strain gave rise to a polybasic FURIN cleavage site which increased its virulence. A recent study also suggested that ins214EPE in Omicron could increase its transmissibility by enhancing sialic-acid receptor binding.
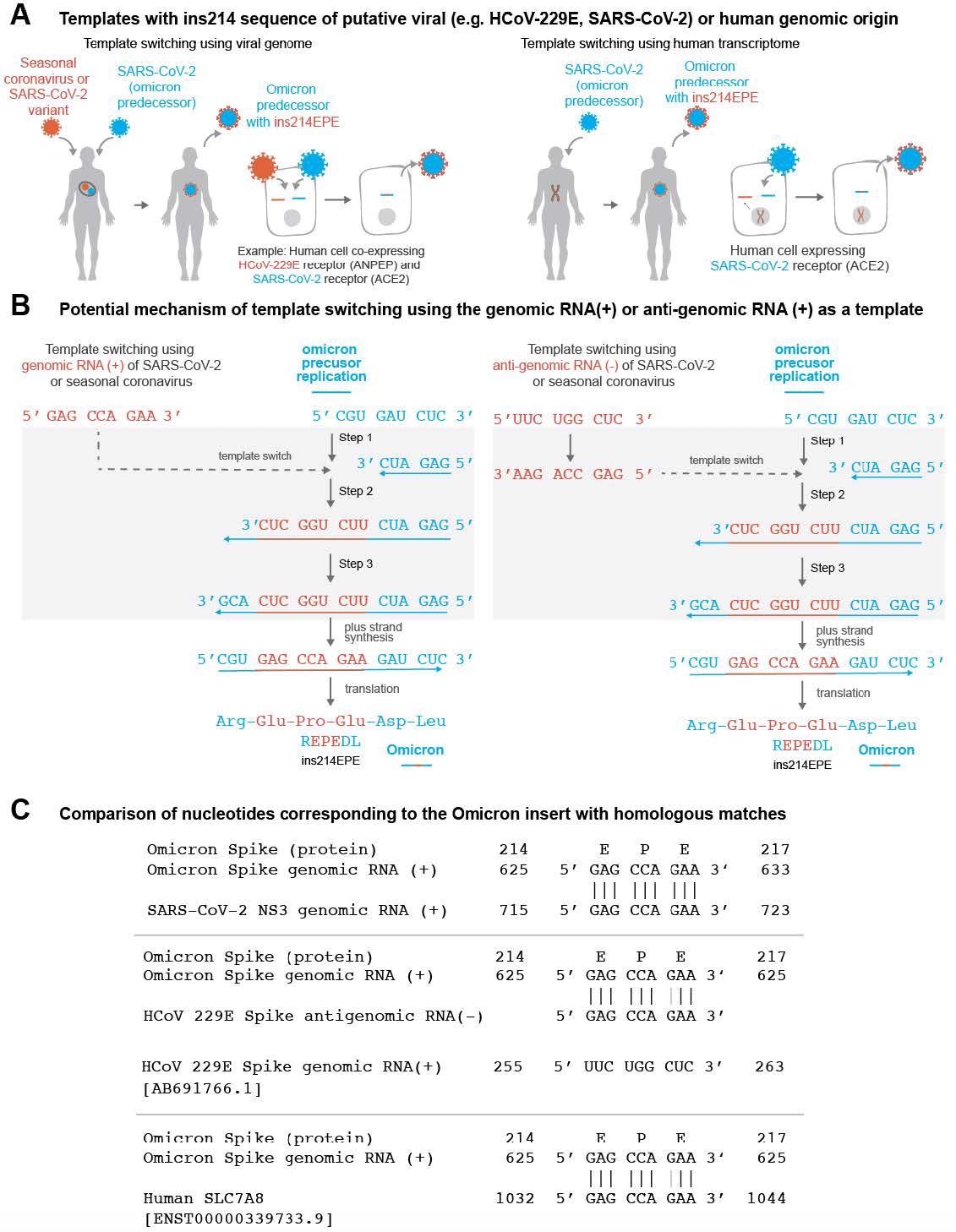
(A) Schematic representation of Omicron evolution through template switching involving viral (e.g. seasonal coronavirus or SARS-CoV-2) or human RNA. (B) Potential mechanism of template switching using viral genomic RNA (positive sense) or anti-genomic RNA (negative sense) as a template. Step 1: Negative strand synthesis begins using Omicron predecessor’s genomic RNA as a template. Step 2: Negative strand synthesis temporarily uses the genomic or anti-genomic RNA of SARS-CoV-2 or a co-infecting virus. Step 3: Negative strand synthesis resumes using Omicron predecessor’s genomic RNA as a template. (C) Examples of matches identical to the nucleotide sequence ‘GAG CCA GAA’ in the SARS-CoV- 2 genome, the HCoV-229E anti-genome, and a human SLC7A8 transcript are shown.
Conclusions
To summarize, Omicron VOC had a unique, never previously observed ins214EPE mutation in its S protein. Although the authors could not conclusively determine the mechanism that gave rise to ins214EPE, they found template switching as the most plausible mechanism for its acquisition. They also suggested that the template-switching event that helped Omicron acquire this mutation did not require a high degree of local homology. Furthermore, the authors highlighted several sources of the template for this insertion, including the SARS-CoV-2 genome, other human CoVs, and human proteins.
To conclude, it is vital to understand the origin and functional consequences of novel mutations that distinguish Omicron from prior SARS-CoV-2 VOCs and VOIs. Hence, the authors emphasized the need for sequencing SARS-CoV-2 genomes from immunocompromised patients for cues leading to the evolution of new SARS-CoV-2 variants.
*Important notice
medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- On the origin of Omicron's unique Spike gene insertion, AJ Venkatakrishnan, Praveen Anand, Patrick J Lenehan, Rohit Suratekar, Bharathwaj Raghunathan, Michiel JM Niesen, Venky Soundararajan, medRxiv pre-print 2022, DOI: https://doi.org/10.1101/2022.06.03.22275976, https://www.medrxiv.org/content/10.1101/2022.06.03.22275976v1
Posted in: Molecular & Structural Biology | Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Biotechnology, Cell, Coronavirus, Coronavirus Disease COVID-19, Epidemiology, Evolution, Gene, Genome, Genomic, immunity, Influenza, Membrane, Mutation, Nucleotide, Nucleotides, Omicron, Phenotype, Polymerase, Polymerase Chain Reaction, Protein, Receptor, Research, Respiratory, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, T-Cell, Transcription, Uracil, Virus

Written by
Neha Mathur
Neha is a digital marketing professional based in Gurugram, India. She has a Master’s degree from the University of Rajasthan with a specialization in Biotechnology in 2008. She has experience in pre-clinical research as part of her research project in The Department of Toxicology at the prestigious Central Drug Research Institute (CDRI), Lucknow, India. She also holds a certification in C++ programming.
Source: Read Full Article