https://bestallergymedicinehq.org/top/how-mindful-breathing-might-actually-help-your-run/
kamagra vs eriacta
Ichthyosis vulgaris (IV)
X-linked recessive ichthyosis (XLRI)
Autosomal recessive congenital ichthyosis
Keratinopathic ichthyosis
Other rare forms
References
Further reading
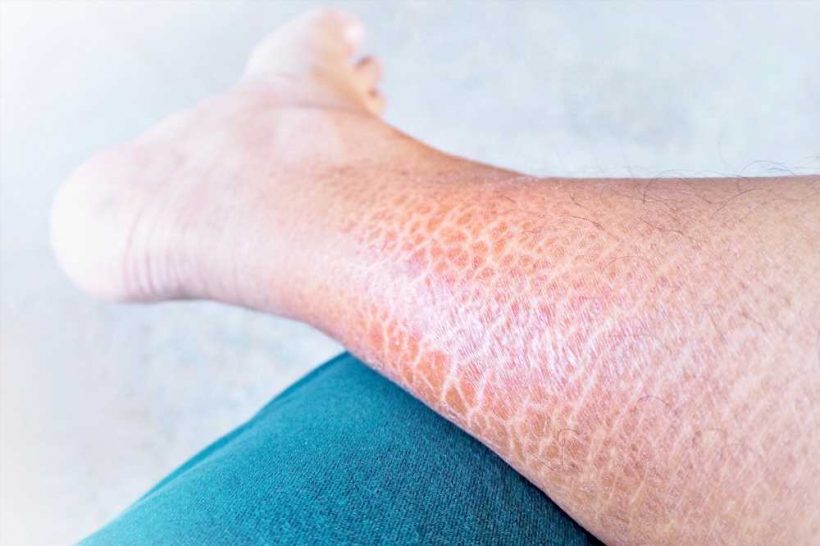
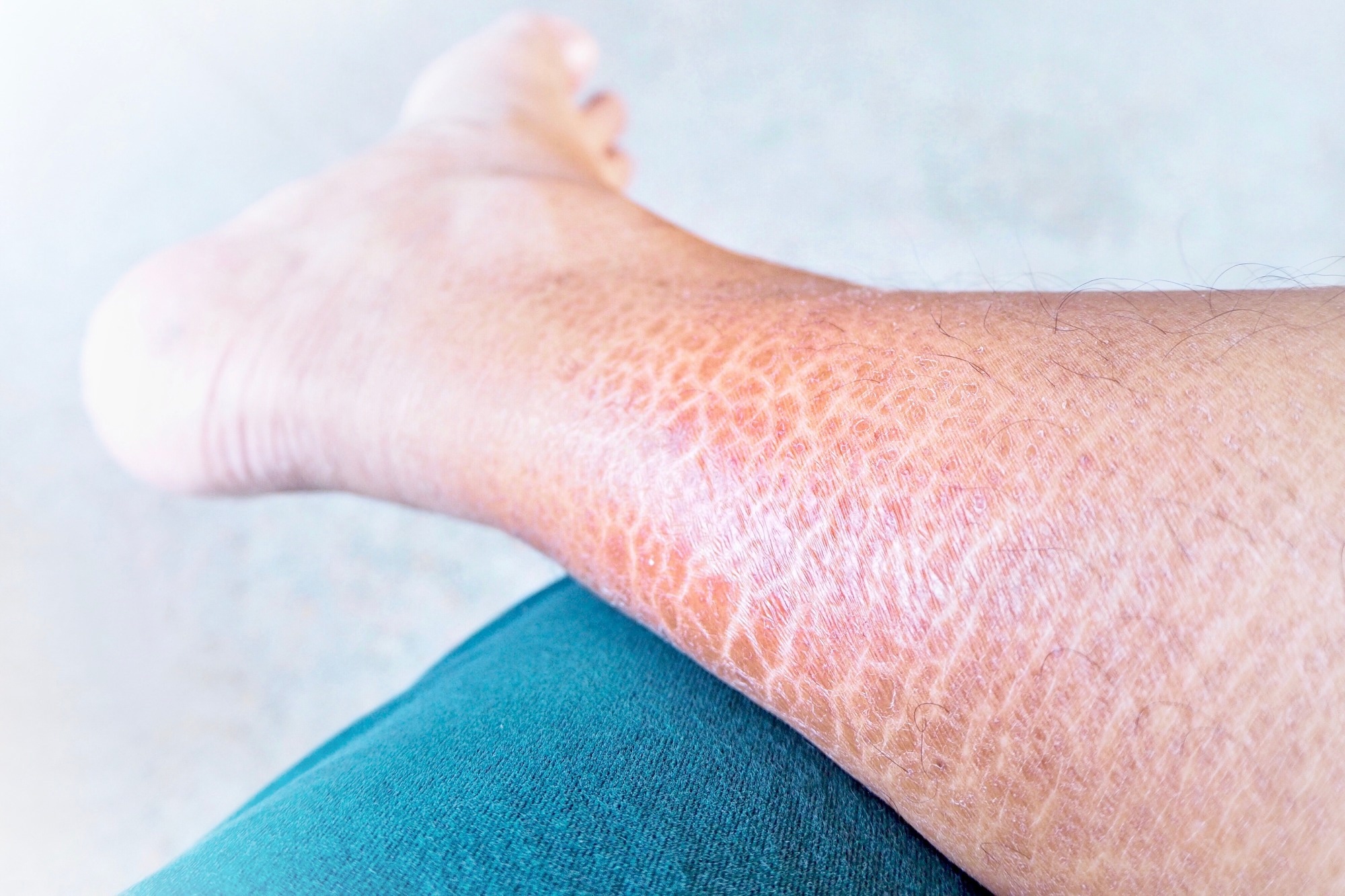
Ichthyosis is an inherited group of skin disorders characterized by xerosis and scaling. Other common phenotypic characteristics are hyperkeratosis, keratosis pilaris, palmar, and plantar hyperlinearity. It is also known as fish scale disease or the disorders of keratinization (DOK).
The clinical symptoms often appear at birth or within the first few years of life. The Ichthyosis Consensus Conference produced a consensus classification for ichthyosis in 2009 based on pathophysiology, clinical symptoms, and manner of inheritance.
It is classified into two types according to this naming system: nonsyndromic forms (clinical symptoms limited to the skin) and syndromic forms (involvement of other organ systems). Ichthyosis Vulgaris and X-linked recessive ichthyosis are the most frequent, both caused by well-known genetic abnormalities.
Depending on the severity of the ichthyosis, treatment should include hydration and lubrication, as well as keratolytic and keratinocyte differentiation modulators. Although oral retinoids are not normally required in the care of common ichthyoses, they are a staple in the systemic management of severe disease.
The majority of ichthyosis treatments try to increase the skin's barrier function. Bathing and carefully applying creams and ointments are important components of an ichthyosis patient's daily regimen.
Ichthyosis vulgaris (IV)
The most prevalent form of ichthyosis is Ichthyosis Vulgaris (IV), which has an incidence of 1:250 to 1:1000. IV is also the mildest form of inherited nonsyndromic ichthyosis, with xerosis, scaling, eczema, and pruritus, and it is strongly linked to atopic symptoms.
The phenotypic manifestations usually arise around an age of 2 months and improve in the summer. The extensor sides of the lower legs and the back are usually afflicted; the chest and abdomen are rarely impacted. Keratosis pilaris and palmoplantar hyperlinearity are both common IV complications.
IV is caused by autosomal dominant mutations in the filaggrin gene (FLG), which is required for epidermal development and skin barrier formation. Population-specific FLG variants of IV have been found in Europeans, Asians, and Africans.
Patients with IVs are more likely to develop atopic dermatitis, asthma, and allergies. This increased risk is likely due to a breakdown in barrier function, which may allow possible allergens to penetrate deeper into the epidermis.
X-linked recessive ichthyosis (XLRI)
XLRI is the second most frequent form of hereditary ichthyosis, with a male incidence ranging from 1:2000 to 1:6000. Clinical manifestations of XLRI are frequently indistinguishable from those of IV.
Symptoms often begin in the neonatal period as generalized desquamation and xerosis, progressing to fine scaling of the trunk and extremities in infancy. Patients develop a brownish, polygonal, plate-like scale that tightly adheres to the skin over time.
Mutations in the STS gene, which codes for steroid sulfatase, on the X chromosome cause XLRI. Some mild and severe XLRI phenotypes may be clinically difficult to distinguish from IV and ARCI (autosomal recessive congenital ichthyosis), respectively.
Because around 90% of XLRI patients have substantial deletions, including STS and surrounding DNA, with contiguous gene loss in certain cases, fluorescence in situ hybridization (FISH) analysis is a valuable tool for detecting XLRI patients and carriers who have such deletions. Nonetheless, while FISH is useful in these circumstances, it is not useful in detecting other persons with partial deletions or point mutations.
Autosomal recessive congenital ichthyosis
ARCI refers to a collection of genetically and phenotypically diverse illnesses that includes harlequin ichthyosis (HI), lamellar ichthyosis (LI), and congenital ichthyosiform erythroderma (CIE). The incidence of ARCI has been estimated to be one in every 200,000 births.
Loss-of-function mutations in ABCA12, which encodes an ATP-binding cassette (ABC) transporter, induce HI. ABCA12 is required for lipid transport into lamellar granules and is important in cornification and lipid barrier formation. Interestingly, while homozygous loss-of-function mutations in ABCA12 result in HI, missense mutations in ABCA12 result in milder LI/CIE symptoms.
LI and CIE can be caused by mutations in one of nine genes – TGM1, NIPAL4/ICHTHYIN, ALOX12B, ALOXE3, CYP4F22, ABCA12, PNPLA1, CERS3, and LIPN16. TGM1 mutations are the most frequent and account for roughly 32% of ARCI17 heritability. A CIE baby is frequently born as a collodion baby. Erythroderma and scaling emerge after the collodion membrane is removed.
The scales in CIE are often fine and white or light grey. The erythroderma in severe cases of CIE is systemic and permanent. However, erythroderma improves in childhood, especially in milder forms.
Another minor form of ARCI is bathing suit ichthyosis (BSI). It is distinguished by a distinct pattern of lesions on the trunk, the most proximal sections of the upper limbs, the scalp, and the neck, but not on the central face and extremities. In South Africa, the term "bathing suit" ichthyosis was introduced to describe this peculiar phenotypic of lamellar ichthyosis. TGM1 missense mutations have been found in 20 BSI cases.
Neonates with HI have thick, armor-like scales on their skin, as well as significant ectropion (eversion of the eyelids), eclabium (eversion of the lips), and ear flattening. Some neonatal HI patients die; however, survival has improved with advances in neonatal intensive care and early treatment with systemic retinoids.
Retinoids can help patients with lamellar ichthyosis, epidermolytic hyperkeratosis, or congenital ichthyosiform erythroderma because the drugs' keratolytic properties allow shedding and prevent future hyperproliferation. It is recommended that these medications be provided in low, effective doses because their use in ichthyosis patients could be lifelong.
Ichthyosis Vulgaris | Causes, Signs & Symptoms, Diagnosis, Treatment
Keratinopathic ichthyosis
Keratinopathic ichthyosis refers to disorders caused by gene mutations in the keratin family. Epidermolytic ichthyosis, or EI, is the most common type. Minor varieties include superficial EI (SEI), annular EI (AEI), and Curth-Macklin ichthyosis. Mutations in the keratin family genes KRT1, KRT2, and KRT10 cause all kinds of keratinopathic ichthyosis.
EI is the most common keratinopathic ichthyotic phenotype, with extensive blister formation and numerous erosions with erythroderma. At birth, the patients have blistering and erythema, which fade with age, and in adulthood, they have extensive epidermolytic hyperkeratosis. Because there is so much phenotypic heterogeneity in EI, baby patients may be clinically difficult to distinguish from other skin conditions.
The term "superficial epidermolytic ichthyosis" was coined for the well-defined condition ichthyosis bullosa of Siemens, which shows a more superficial pattern of epidermolysis than EI. Mutations in KRT2 cause it, and clinical symptoms include minor epidermal hyperkeratosis across flexural areas, blister formation, and the development of superficially denuded areas of hyperkeratotic skin.
Annular epidermolytic ichthyosis is a unique phenotype of EI. It is distinguished by the sporadic appearance of annular, polycyclic, erythematous, scaly plaques over the proximal extremities and trunk. It is caused by a specific mutation in KRT10 that swaps an alanine with a proline at residue 12.
Rare autosomal recessive mutations in KRT10 cause autosomal recessive epidermolytic ichthyosis. AREI cases are caused by nonsense mutations that result in a premature termination codon of KRT10, resulting in the protein's complete absence.
Another uncommon disorder is Ichthyosis Curth-Macklin, caused by autosomal dominant mutations in KRT1. It is distinguished by widespread, spiky, or verrucous hyperkeratosis of the big joints and trunk, with or without palmoplantar keratoderma.
Congenital reticular ichthyosiform erythroderma, also known as ichthyosis with confetti (IWC), is an extremely unusual skin condition in which patients are born with erythroderma on nearly the whole body surface due to faulty skin barrier function, prominent scales, and palmoplantar keratoderma.
Hundreds to thousands of pale confetti-like specks occur on the body surface early in life, increasing in quantity and size with maturity. Histological findings include epidermal thickening in ichthyotic skin and keratinocyte differentiation disruption with parakeratosis.
Other rare forms
Loricrin keratoderma (LK) is a genodermatosis with mild ichthyosis that runs in families. LK is frequently connected with pseudoainhum; affected individuals are occasionally born as collodion babies.
LOR, which encodes loricrin, a key component of the cornified cell membrane present in terminally developed epidermal keratinocytes, is mutated in LK.
Erythrokeratoderma variabilis (EKV) is an autosomal dominant condition usually manifest in the first year of life. The two most common skin symptoms are localized, sharply confined keratotic lesions and migrating erythematous patches.
Mutations in GJB3 and GJB4, which encode connexins 31 and 30.3, respectively, have been found in EKV. There is presently no EKV-specific therapy available.
References
- Dorf IL, Sommerlund M, & Koppelhus U (2020). Ugeskrift for Laeger, 182(17), V10190611. doi:10.1186/s13023-015-0336-4. https://ojrd.biomedcentral.com/articles/10.1186/s13023-015-0336-4
- Takeichi T, & Akiyama M (2016). Inherited ichthyosis: Nonsyndromic forms. The Journal of Dermatology, 43(3), 242–251. doi:10.1111/1346-8138.13243. https://onlinelibrary.wiley.com/doi/10.1111/1346-8138.13243
- Marukian NV, & Choate KA (2016). Recent Advances in Understanding Ichthyosis Pathogenesis. F1000Research, 5, F1000 Faculty Rev-1497. doi:10.12688/f1000research.8584.1. https://f1000research.com/articles/5-1497/v1
- Limmer AL, Nwannunu CE, Patel RR, et al. (2020). Management of Ichthyosis: A Brief Review. Skin Therapy Letter, 25(1), 5–7. https://pubmed.ncbi.nlm.nih.gov/32023022/
- Guerra L, Diociaiuti A, El Hachem M, et al. (2015). Ichthyosis with confetti: clinics, molecular genetics and management. Orphanet Journal of Rare Diseases, 10, 115. doi:10.1186/s13023-015-0336-4. https://ojrd.biomedcentral.com/articles/10.1186/s13023-015-0336-4
Further Reading
- All Skin Content
- Cutaneous Manifestations of Internal Malignancy
- What is Subcutaneous Tissue?
- Milia in Babies
- Why Does Skin Go Wrinkly in Water?
More…
Last Updated: Sep 5, 2023
Source: Read Full Article